
La cétolyse est l’unique source d’Acétyl CoA mitochondrial des cellules tumorales, l’inhibition de l’enzyme Succinyl CoA-acétoacétyl-CoA transférase, devrait les rendre vulnérables.
Maurice Israël
2 av. Aristide Briand 91440 Bures sur Yvette et CNRS 91190 Gif surYvette, France.
Résumé
Les Cellules tumorales ont un métabolisme hybride anabolic/catabolic: certaines enzymes sont phosphorylées comme si des hormones cataboliques ( glucagon epinéphrine) avaient activé des récepteurs couplés à des protéines Gs, tandis que d’autres enzymes sont dans une configuration anabolique, induite par des récepteurs de type tyrosine kinase stimulés par l’insuline. Les cellules tumorales seront par conséquent contraintes de câbler autrement leur métabolisme. Ceci leur confère un avantage sur des cellules différentiées, qui répondent préférentiellement aux signaux cataboliques et mobilisent leurs réserves, au profit des cellules tumorales. Le commutateur de sélection pancréatique entre anabolisme et catabolisme dépend du GABA, sa défaillance expliquerait la double libération d’hormones cataboliques et anaboliques, ainsi qu’une désensibilisation progressive des récepteurs de l’insuline dans les cellules différenciées. Par contre, des nouvelles cellules souches dont les récepteurs à l’insuline n’ont pas été désensibilisés, répondront aux signaux anaboliques et cataboliques et câbleront leurs voies métaboliques sur le mode tumoral. Puisque ces cellules mitotiques doivent obligatoirement synthétiser des acides gras, pour leurs nouvelles membranes, il y aura un blocage automatique de leur dégradation en acétyl CoA ; puisque la Pyruvate kinase M2 et la Pyruvate deshydrogénase des cellules tumorales sont inhibées, la source glycolytique d’acétyl coA est également close. Les cellules tumorales doivent alors obligatoirement convertir des corps cétoniques en acétyl CoA mitochondrial, inhiber cette cétolyse pourrait les détruire ou les contraindre à normaliser leur métabolisme.
Introduction
Dans des publications antérieures (1,2) le métabolisme du cancer, ainsi que les signaux qui contrôlent ce métabolisme, ont été décrits pour mettre en évidence « le câblage métabolique » si particulier des cellules tumorales. Schématiquement, dans les cellules tumorales, certaines enzymes sont phosphorylées et inhibées, tel qu’il est observé lorsque des hormones cataboliques induisent une gluconéogenèse, tandis que d’autres enzymes, dé-phosphorylées, semblent répondre aux hormones anaboliques, induisant : une glycolyse, une synthèse de glycogène avec inhibition de la lyse du glycogène, et des synthèses protéiques et lipidiques. Cette situation hybride des enzymes des cellules tumorales entraine un nouveau câblage des voies métaboliques de ces cellules. Ceci leur confère alors un avantage métabolique, qui leur permet de piller les réserves des autres tissus de l’organisme, devenus sélectivement sensibles aux hormones cataboliques et résistants à l’insuline (3,4). On sait que la sélection entre anabolisme et catabolisme est l’une des fonctions essentielles du pancréas endocrine. Il était donc naturel d’incriminer cette fonction pour expliquer l’état hybride observé pour les enzymes des cellules tumorales, ainsi que la réponse préférentiellement catabolique de cellules différentiées devenues résistantes à l’insuline.
Normalement, les cellules bêta du pancréas sécrètent parallèlement à l’insuline, un médiateur inhibiteur : l’acide gamma aminobutyrique (GABA) qui met au repos les cellules voisines du pancréas endocrine, les cellules alpha qui libèrent le glucagon et les cellules delta qui libèrent la somatostatine. Ainsi lorsque l’insuline anabolisante est libérée, le glucagon et son action catabolique sont mis au repos par le GABA. Il a été récemment suggéré que c’est l’altération de cette régulation fondamentale du pancréas endocrine qui explique la configuration hybride des enzymes des cellules tumorales, dont certaines répondent aux hormones cataboliques et d’autres aux hormones anaboliques. C’est cette situation qui conduit à un câblage métabolique spécifique des cellules tumorales (2). Alors que les cellules différenciées de l’organisme semblent répondre préférentiellement aux hormones cataboliques, qui mobilisent les réserves de l’organisme au profit de la tumeur. Nous allons dans ce qui suit expliquer cette situation métabolique qui pourrait être à l’origine de l’état cancéreux.
Effets des hormones cataboliques et anaboliques
On sait que la mobilisation des réserves tissulaires est une situation physiologique, associée à l’hypoglycémie ; dans ce cas, les hormones cataboliques, glucagon et norépinephrine, agissent sur des récepteurs couplés aux protéines G, de type Gs, qui stimulent l’adénylate cyclase et la synthèse d’AMP cyclique. Celle-ci va ensuite stimuler des protéines kinases (PKA/Src), qui phosphorylent in fine des enzymes en aval. Le résultat de ces phosphorylations est la production de glucose par lyse du glycogène (glycogénolyse) et par synthèse de glucose (néoglucogenèse), tandis que d’autres enzymes activées par phosphorylation vont mobiliser les acides gras pour produire des corps cétoniques. Lors du catabolisme les enzymes pyruvate kinase (PK) et pyruvate déshydrogénase (PDH) sont phosphorylées et inactives, ce qui ferme l’entrée du cycle de Krebs, et épargne l’oxaloacétate (OAA) à l’origine de la voie de synthèse du glucose. Glucose et corps cétoniques sont les nutriments indispensables au fonctionnement d’autres tissus comme les muscles ou le cerveau. On voit donc que la phosphorylation de PK et PDH indique bien l’action catabolique, associée à la néoglucogénèse.
A l’inverse, lorsque la nourriture est abondante, une augmentation de la glycémie provoque la libération d’insuline et d’autres hormones anabolisantes. Dans l’anabolisme, le métabolisme glycolytique se remet en route, PK et PDH sont déphosphorylées via l’activation de phosphatases et l’inhibition des protéines kinases de ces enzymes. On sait aussi que l’augmentation d’un activateur de glycolyse : le fructose 2-6 bis phosphate, stimule une enzyme la phosphofructokinase dans la direction glycolytique, et ferme la néoglucogenèse. L’action de l’insuline et d’autres hormones anabolisantes est transmise par des récepteurs de type tyrosine kinase (Tyr kin R) qui vont activer deux voies de signalisation : les MAP kinases et PI3 kinase, celles-ci contrôlent mitose et survie cellulaire. Cet état anabolique est associé à la synthèse des substances nécessaires au renouvellement des tissus, les cellules souches entrent en mitose. L’activation des récepteurs de l’insuline va aussi changer le métabolisme en déphosphorylant une série d’enzymes ; comme la PK et la PDH, il y aura activation de phosphatases et inhibition des protéines kinases pour les différentes enzymes. Cet effet des récepteurs (Tyr kin R) de l’insuline, est transmis à une première kinase (PKB), qui inhibe à ensuite les kinases des différentes enzymes, qui seront in fine, déphosphorylés. Le résultat métabolique est une augmentation de l’entrée du glucose (incorporation du transporteur de glucose à la membrane cellulaire) ce qui stimule la glycolyse. Il y aura aussi une activation des synthèses de glycogène, de protéines, d’acides gras et lipides, nécessaires aux nouvelles cellules. Les deux états opposés que nous avons schématisés s’excluent mutuellement par le biais d’une régulation fondamentale de pancréas endocrine, un commutateur de sélection dépendant du GABA. D’autres substances (Zn 2+, choline) secrétés par la cellule bêta parallèlement au GABA et à l’insuline, sont également impliquées dans cette régulation.
Commutateur de sélection métabolique contrôlé par le GABA
Le commutateur de sélection entre anabolisme ou catabolisme est situé dans le pancréas endocrine. Lorsque les cellules bêta libèrent l’insuline anabolisante en réponse à une glycémie élevée, elles inhibent par une libération parallèle de GABA, les cellules alpha libérant le glucagon et inhibent ainsi le catabolisme. Le GABA bloque également les cellules delta qui habituellement libèrent la somatostatine. Ceci renforce l’action de l’insuline, puisque l’hormone de croissance et l’IGF (insuline like growth factor) ne sont plus inhibés par la somatostatine. Le GABA inhibe efficacement les cellules alpha et delta via des récepteurs ionotropiques de type GABA A. L’activation de ces récepteurs déclenche une entrée de Cl – qui hyper-polarise les cellules. Cette hyperpolarisation bloque l’influx de Ca2+ ce qui inhibe la libération de glucagon et de somatostatine.
Quelles seraient les conséquences d’une défaillance de ce système dépendant du GABA ?
Cela peut se produire si l’enzyme glutamate décarboxylase (GAD) synthétisant le GABA, est inhibée, ou s’il y a un déficit de vitamine B6 le cofacteur GAD, (On sait aussi que des autoanticorps anti GAD peuvent provoquer un diabète de type I en détruisant les cellules bêta), des pesticides peuvent aussi bloquer la synthèse et le métabolisme du GABA.
Une libération de GABA défaillante, ferait que la cellule bêta serait incapable d’inhiber la libération du glucagon et le catabolisme, tandis que l’insuline anabolisante serait libérée. Un message hybride catabolique / anabolique serait alors envoyé aux tissus ; et en effet, les cellules tumorales ont une réponse mixte. Ceci est illustré par le blocage de PK et PDH par phosphorylation comme dans une situation catabolique avec gluconéogenèse. Cependant, ceci est paradoxalement associé à une glycolyse très active et a une forte condensation en citrate à l’entrée du cycle de Krebs. En outre les synthèses dans la cellule tumorale sont élevées comme dans l’anabolisme. Pour surmonter le blocage de PK et PDH, les cellules tumorales doivent alors « rebrancher » autrement le système, elles devront utiliser pour la réaction de condensation en citrate, l’acétyl CoA provenant de corps cétoniques, formés dans le foie à partir des réserves lipidiques et d’acide gras, et de l’oxaloacétate (OAA) formée via la phosphoénolpyruvate carboxykinase ou la malate déshydrogénase. En aval de la réaction de condensation entre l’OAA et l’acétyl CoA en citrate, un nouveau « stop » dans le cycle de Krebs favorise l’efflux du citrate mitochondrial vers le cytosol. Ce flux alimente la synthèse des acides gras, nécessaires pour constituer les membranes lipidiques des cellules mitotiques, via l’ATP citrate lyase ; l’acétyl CoA carboxylase et fatty acid synthetase (FAS), enzyme de synthèse des acides gras (1-4).
Les enzymes des cellules différenciées, hépatocytes ou adipocytes, n’adoptent pas cette configuration hybride catabolique / anabolique, comme si ces cellules étaient relativement résistantes à l’insuline. C’est une situation qui rappelle le diabète de type 2, associé à une désensibilisation des récepteurs de l’insuline, intériorisés par endocytose avant protéolyse. Cette désensibilisation intervient lorsque les cellules sont soumises de manière chronique à de l’insuline. Pour éviter cette désensibilisation, les cellules bêta qui ont libéré l’insuline doivent alors être capables d’arrêter complétement sa libération et mettre fin à son action. Dans ce cas, le GABA libéré agit sur des autorécepteurs des cellules bêta, des récepteurs métabotropiques de type GABA B, qui sont couplés à des protéines de type Gi (qui inhibent l’adénylate cyclase). Ceci fermera après plusieurs étapes, le mécanisme de libération de l’insuline (5). De plus, l’activation des récepteurs GABA B semble augmenter l’incorporation dans la membrane de canaux potassium (KATP) inhibés par l’ATP. L’augmentation de la conductance membranaire au potassium (6) et de l’efflux de potassium qui s’en suit, hyper polarise la cellule et arrête complètement la libération d’insuline. Par conséquent, la libération de GABA des cellules bêta fonctionne comme un auto-inhibiteur (autocrine), éteignant la libération d’insuline. Si la libération de GABA est déficiente il y aura alors une fuite persistante de la libération d’insuline, qui à la longue, désensibilisera les récepteurs de l’insuline sur des cellules différenciées (Rappelant le diabète de type 2) ces cellules pourront toutefois répondre aux signaux cataboliques. Dans les cellules tumorales, la situation est très différente, puisqu’elles répondent à l’insuline et au glucagon, elles adoptent alors un métabolisme hybride, anabolique / catabolique. L’explication la plus simple est que contrairement aux cellules différenciées soumises à un processus de désensibilisation prolongé de leurs récepteurs d’insuline ; les nouvelles cellules souches expriment de nouveaux récepteurs d’insuline, qui n’ont pas été soumis à cette lente désensibilisation; et puisque ces cellules sont également pourvues de récepteurs au glucagon ou à l’épinéphrine de type Gs, elles auront un métabolisme hybride anabolique / catabolique qui reconfigure les voies métabolique (Figure 1) .
En outre, dans la médullosurrénale, une défaillance de GABA stimulera la libération de norépinephrine, qui inhibera la libération de la somatostatine et renforcera l’action de l’hormone de croissance et de l’IGF. Nous verrons que l’hormone de croissance stimule l’enzyme adipocyte triglycéride lipase (ATGL) et la production de Diacyl glycérol (DAG). Le glucagon stimule également la libération de cortisol, induisant la protéolyse musculaire pour fournir des aminoacides aux synthèses protéiques des cellules tumorales.
En somme
Les trois hormones cataboliques sont opérationnelles, mobilisant des réserves de l’organisme au profit de cellules mitotiques qui ont une réponse métabolique hybride-reconfigurée. Le métabolisme du cancer serait alors la conséquence d’une défaillance de GABA, supprimant un mécanisme de contrôle essentiel du pancréas endocrine. Surveiller cette fonction du pancréas endocrine dépendante du GABA, de sa synthèse et du cofacteur de l’enzyme de synthèse, la vitamine B6, pourraient jouer un rôle dans la prévention du cancer. Corriger les conséquences métaboliques de cette altération du pancréas endocrine, pourrait également aider l’approche thérapeutique.
L’exception musculaire
Il y a cependant une exception aux effets du glucagon, ou de l’épinephrine, dans les muscles striés. Ici l’hormone catabolique ne déclenche aucune synthèse de glucose par neoglucogenesis, alors que la glycogénolyse et la protéolyse sont activées. Le retour de l’activité glycolytique et de la glycolyse oxydative, semblent être liés à la fonction même des muscles striés, qui doivent être prêts pour échapper par la fuite aux prédateurs. Comment fonctionne cette exception ? Lors de la stimulation musculaire la dépolarisation de la membrane génère un mouvement de charges le long des tubules de T jusqu’aux récepteurs de ryanodine des sacs de réticulum endoplasmique ; ce qui entraine une libération de calcium dans le cytosol, ce qui initie la contraction. La libération de calcium active également une phosphodiesterase qui hydrolyse le cAMP. La diminution de cAMP lève son inhibition sur la synthèse du fructose 2-6 bis phosphate, qui est l’activateur allostérique de la phosphofructokinase et de la glycolyse. La libération de calcium dans le cytosol stimule
également une phosphatase, la calcineurine PP2B, qui déphosphoryle un inhibiteur (Inhibiteur I1) de la phosphatase PP1. L’activation de PP1 pourrait alors activer d’autres phosphatases (des tyrosines phosphatases). Qui vont alors activer PK (forme M1) et la PDH et ouvrir la source glycolytique de l’ acétyl CoA à l’entrée du cycle Krebs. En outre, la myoglobine musculaire tirera l’oxygène nécessaire pour oxyder le NADH, et déclencher la réaction de condensation en citrate. Nous verrons que les cellules tumorales vont via le calcium intracellulaire activer la phosphodiesterase et l’hydrolyse de l’AMP cyclique déclenchant une augmentation de l’entrée du glucose et de la glycolyse mais que la PK (sa forme M2), reste bloqué par phosphorylation sous forme dimérique. La PDH reste aussi fermée comme si in autre inhibiteur de PP1 avait annulé l’effet de la calcineurine, nous verrons que c’est l’élévation de DAG qui active la protéine kinase C (PKC) et entraine la formation de l’inhibiteur CPI 17 qui ferme la fin de la glycolyse et la source glycolytique de l’acétyl CoA. Cet effet de DAG sur la PKC est comparable à l’action carcinogène des esters de phorbol via PKC.
Sélection de la source d’acétyl-CoA
L’action de l’insuline active en aval de son récepteur, une phospholipase qui produit de l’IP3 (Inositol3,4,5 Phosphate) et du Diacylglycerol (DAG). L’augmentation de IP3 va entrainer une libération du calcium du réticulum dans le cytosol qui active comme pour l’exception musculaire, une phosphodiesterase diminuant cAMP, ce calcium stimule aussi la calcineurine phosphatase, qui inactive l’inhibiteur de PP1. Les phosphatases PP1, PP2B, pourront alors déphosphoryler et activer PK et PDH et ouvrir la source glycolytique de l’acétyle CoA. Mais ici la libération du calcium via IP3 est associée à la formation du diacylglycérol (DAG). Cette «augmentation métabolique» du calcium, diffère de l’augmentation musculaire du calcium suscitée par l’activité électrique (sans produire DAG). Dans le cas de l’insuline, le DAG formé permettra de sélectionner la source de l’acétyl CoA. Une élévation de DAG, va stimuler la protéine Kinase C (PKC) qui induit la formation d’un autre inhibiteur (CPI-17) de la phosphatase PP1. L’inhibition de PP1 (une phosphatase sérine) entraine l’inhibition d’autres tyrosine phosphatases, et va bloquer ainsi le l’activation de PK et PDH par déphosphorylation, et fermer la source glycolytique de l’acétyl CoA. C’est alors la beta oxidation mitochondriale des acides gras qui devra fournir l’acétyl CoA. Dans cette situation, AMP est encore élevé, et active AMP kinase, qui est un inhibiteur de l’acétyl CoA carboxylase (ACC), et de la synthèse des acides gras ; en effet, le malonyl CoA qui n’est plus produit, est un inhibiteur du transporteur- carnitine des acides gras, puisqu’il est au plus bas, la dégradation des acides gras en acétyl CoA est sera activée, ouvrant la source lipidique d’acétyl CoA. Mais lorsque l’effet anabolisant de l’insuline prend le dessus, l’ACC est stimulée et formera du malonyl CoA, qui inhibera le transport des acides gras dans la mitochondrie et de leur dégradation par beta oxidation, ce qui fermera la source lipidique de l’acétyl CoA. La synthèse des acides gras, des lipides et membranes, prend le dessus. L’activation de PKC par DAG va ensuite stimuler une désaminase de l’AMP et la 5’nucléotidase, entrainant une diminution progressive de l’AMP. La stimulation de l’AMP kinase est stoppée ce qui lève l’inhibition de l’ACC, ouvrant alors la voie de synthèse pour les acides gras. Dans ce cas, le citrate quitte les mitochondries, redonne l’acétyl CoA dans le cytosol, via l’ATP citrate lyase, puis le malonyle- CoA via l’ACC, jusqu’à l’enzyme de synthèse des acides gras (FAS). Le malonyl-CoA formé par l’ACC inhibera le transporteur à carnityl des d’acide gras de la mitochondrie, et fermera leur bêta oxydation et la source d’acide gras pour l’acétyl CoA. Il sera donc nécessaire d’ouvrir à nouveau la source glycolytique de l’acétyl CoA et de déphosphoryler PK et PDH. C’est vraisemblablement la diminution du DAG, consommé pour former des triglycérides, qui annulera la stimulation de PKC par DAG, et la formation de CPI-17, l’inhibiteur PP1. Ceci va alors permettre la déphosphorylation de PDH et PK et ouvrir à nouveau la source glycolytique de l’acétyl CoA. Cette situation est sans doute en relation avec les travaux de Randal décrivant les interactions réciproques entre le glucose et le métabolisme des acides gras.
Dans les cellules tumorales qui synthétisent leurs acides gras et lipides et qui ont de ce fait bloqué via le malonyl CoA, la dégradation des acides gras, il serait nécessaire d’ouvrir la source glycolytique de l’acétyl CoA, mais cette dernière étape n’est plus opérationnelle. Comme si les cellules tumorales ne parvenaient pas à faire baisser le taux de DAG, malgré sa conversion en triglycérides, comme s’il y avait une production accrue de DAG. Il est probable que c’est l’hormone de croissance activée par la baisse de somatostatine du pancréas qui est à l’origine de cette surproduction de DAG (discutée ci-dessus) car l’hormone de croissance stimule l’adipocyte triglycéride lipase productrice de DAG. Avec les 2 sources d’acétyl CoA (acide gras ou glycolytique) fermées, il ne reste plus à la cellule tumorale que les corps cétoniques pour couvrir sa demande vitale en acétyl CoA mitochondrial. Ces corps cétoniques sont produits dans le foie et autres tissus sélectivement sensibles aux hormones cataboliques et résistants à l’insuline, Ils vont être reconvertis par la cellule tumorale en acétyl CoA ; dans cette voie métabolique l’enzyme succinyl CoA –acetoacetate- CoA transférase (SCOT) joue un rôle primordial, il conviendrait donc de l’inhiber.
La cétogenèse et l’utilisation des corps cétoniques.
Un hépatocyte soumis à l’action du glucagon, est représenté figure 2, il produit du glucose et va synthétiser dans sa mitochondrie des corps cétoniques. Les acides gras y sont transportés par le transporteur à carnitine, et vont y être dégradés par bêta oxydation en acétyl CoA. Quatre enzymes sont impliqués : la thiolase ; l’hydoxy methyl glutaryl CoA synthétase ; l’hydroxymethyl glutarylCoA lyase et la beta hydrohybutyrate deshydrogénase. Elles vont aboutir au bêta hydroxy butyrate, il dérive directement de l’acétoacétate, il est la forme de transport des corps cétoniques, vers d’autres tissus. La cellule tumorale figure 2, va incorporer le bêta hydoxy butyrate où il sera reconverti en acétyl CoA mitochondrial, par un chemin mettant en jeu 3 enzynes : à nouveau la beta hydroxybutyrate deshydrogénase ; puis la succinylCoA acetoacetyl, CoA transférase (SCOT) et à nouveau la thiolase en sens inverse. On voit que c’est SCOT qui est spécifique pour cette reconversion des corps cétoniques en acétyl CoA, qui pourra alors alimenter la réaction de condensation en citrate, qui démarre le cycle de Krebs de la cellule tumorale. Cet apport lui est capital puisque les autres sources d’acétyl CoA, glycolytiques ou à partir des acides gras sont fermées, comme nous l’avons expliqué. Le citrate va ensuite quitter la mitochondrie pour alimenter la synthèse cytosolique des acides gras, lipides et cholestérol, nécessaires pour constituer les nouvelles membranes des cellules mitotiques. Le citrate sorti dans le cytosol sera lysé par l’ATP citrate lyase en acétyl CoA et Oxaloacétate. L’acétyl CoA va alimenter la voie de synthèse des acides gras, alors que l’oxaloacétate va orienter les transaminations associées à l’apport d’amino acides, vers la production de pyruvate à partir d’alanine, et in fine de lactate. Lors de précédents travaux nous avons suggéré d’inhiber l’ATP citrate lyase par l’hydroxycitrate, et ainsi la voie de synthèse des acides gras. Nous avons aussi pensé que l’acide lipoique pourrait réactiver la PDH dont il est le co-facteur, mais il est plus probable que ce réducteur et produit de la PDH agit sur la réaction de condensation en citrate, normalement inhibée par le NADH. En tout cas la combinaison hydroxycitrate et l’acide lipoique a été particulièrement efficace sur des souris porteuses de cancer. La liste des produits publiés, pouvant agir sur le métabolisme tumoral mériterait d’être ré évaluée en trouvant des combinaisons encore plus efficaces.
Ici nous allons centrer le problème sur l’utilisation des corps cétoniques par la tumeur. En inhibant SCOT nous allons encore réduire la synthèse d’acétyl CoA mitochondrial, cet effet sera en amont de l’acide lipoique et de l’hydroxycitrate. Quels sont donc les inhibiteurs de SCOT ? La base de données BRENDA nous donne une liste de produits qui pourraient être très toxiques : dinitrophénylacétate, et autre produits nitrés, dérivés fluorés de l’acide succinique (2,2 difluorosuccinate) (7). On y trouver aussi l’acide succinique il est le produit de la réaction enzymatique, mais aussi le desulfo CoA, desulfopanteteine, le 3-sulfopropanoate, citrate, N-acétylcysteamine, acetylimidazole et d’autre produits peu spécifiques. Les recherches de Picart et Jenks (8), sur le centre actif de l’enzyme SCOT et le complexe enzyme- substrat ont montré que l’acide acétohydroxamic pouvait bloquer la réaction. Or il se trouve que l’acide acetohydroxamic est utilisé en médecine sous forme de tablettes « lithostat » pour traiter des infections de la vessie par des bactéries productrice d’ammoniac, et pouvant provoquer des calculs rénaux. Son effet inhibiteur sur SCOT pourrait donc être utile et avoir des effets anti-cancer. Le salicylhydroxamic, semble avoir des effets similaires sur des infections urinaires. On sait aussi qu’une catégorie d’inhibiteurs d’histone desacétylase (HDAC) sont aussi des dérivés de l’acide hydroxamique citons le suberoylanilide hydroxamic acid SAHA ou la trichostayine A ou le vorinostat, ils pourraient alors bloquer SCOT expliquant les effets anti-cancer qui ont été observés mais attribués au blocage de HDAC, voir par Pal et Saha (9).
Signalons enfin que le dichloroacetate antérieurement utilisé pour bloquer les protéines kinases phosphorylant PK et PDH (10) et qui bloque les cellules tumorales, a aussi un effet inhibiteur sur l’utilisation des corps cétoniques, en inhibant l’incorporation du bêta hydroxybutyrate (11). On peut donc considérer que l’inhibition de SCOT et de l’utilisation des corps cétoniques, privera les mitochondries des cellules tumorales en acétyl CoA , et comme les 2 autres sources (glycolytiques et bêta oxidation) sont fermées, le flux de citrate vers les voies de synthèse lipidiques du cytosol sera ainsi réduit minimum. Cependant les cellules tumorales peuvent encore surmonter le problème, car elles contiennent de l’acétyl CoA synthetase et de l’acetoacetyl CoA synthetase, pouvant alimenter la synthèse des acides gras et cholestérol.
Rôle des acetylCoA synthetase et acetoacetyl CoA synthetase des cellules tumorales.
Les cellules tumorales ont aussi acquis la possibilité d’utiliser directement l’acide acétique pour le convertir en acétyl CoA via l’acétyl CoA synthétase du cytosol, comme le ferait une terminaison neuromusculaire cholinergique, qui syntétisera ensuite de l’acétylcholine via la choline acétyltransférase, alors que les synapses cholinergiques du système nerveux central utilisent préférentiellement le pyruvate comme précurseur de la moitié acétyl de l’acétylcholine(12).
Afin d’éviter que la cellule tumorale puisse réactiver à partir de l’acétyl CoA synthétase cytosolique, la synthèse de l’acétyl CoA et alimenter ainsi la formation des acides gras et lipides membranaires (via l’acétyl CoA carboxylase ACC, et Fatty acid synthetase FAS), il conviendrait d’inhiber son acétyl CoA synthetase. L’allicine est un inhibiteur utilisable (13), également intéressant l’acide orotique (13bis) voir aussi des dérivés de la quinoxaline : 1-(2,3-di(thiophen-2-yl)quinoxalin-6-yl)-3-(2-methoxyethyl)urea, qui a manifestement des effets anti-cancer (14).
Pour ce qui est de l’acetoacetylCoA synthetase elle alimente la synthèse du cholestérol via hydroxy methyl glutarylCoA synthetase, puis une réductase formant du mévalonate, conduisant après plusieurs étapes au cholestérol.
Le point commun entre l’acétyl CoA synthetase et l’acéto acetyl CoA synthétase est que ces 2 enzymes fonctionnent en formant d’abord un intermédiaire acyl – adénylate en présence d’ATP. Or ce type d’enzyme est inhibé par des composés comme le sulfonyladénosine (15) ; ou par des dérivés du Celecoxib ( AR12, AR14) qu’il conviendrait d’essayer (16) .
Enfin, en aval de l’acétyl CoA carboxylase et du malonyl CoA qui ferme, comme on l’a vu, la dégradation mitochondriale des acides gras, un blocage de FAS (Fatty ac. Synthetase) maintiendra le malonyl CoA en amont, pour fermer la bêta oxidation, tout en inhibant la synthèse des acides gras ; les inhibiteurs connus de FAS sont : cerulenin, C75, orlistat (17).
Enfin, en aval de l’acéto acétyl CoA synthétase, sur la voie de synthèse du cholestérol, l’enzyme 3 hydroxy 3 méthyl glutaryl CoA synthétase est classiquement inhibée par les statines ( simvastatinou autre).
L’approche qui a été décrite ici, a pour but d’empêcher le développement de la tumeur en fermant sont unique source d’acétyl CoA mitochondriale : la cétolyse via SCOT ; l’inhibition de cet enzyme par l’acide acetohydroxamic ou ses dérivés sera testée. La synthèse des acides gras, pourra être également inhibée en aval de l’acétyl CoA carboxylase (ACC), par des inhibiteurs de FAS. On réduira l’entrée d’acétyl CoA et acétoacetyl CoA cytosolique (inhibition des acyl synthétases) et de la synthèse du cholestérol, ce qui devrait empêcher la formation des membranes des cellules tumorales.
Une autre approche possible, thérapeutique, ou préventive, pourrait être la réactivation des voies métaboliques bloquées dans les cellules tumorales (voir 18, 19) ou de contrôler les altérations hormonales (régulation GABA du pancréas), qui font que des cellules souches ont reprogrammé leur métabolisme à leur avantage, et cessent de se différencier, formant une tumeur dans l’organe qu’elles devaient réparer.
Summary
Tumor cell display hybrid metabolic features: some enzymes seem phosphorylated, following catabolic hormonal actions, mediated by Gs coupled receptors, while others adopt a configuration found in anabolism, mediated by tyrosine kinase receptors stimulated by insulin. Consequently, tumor cells rewire their metabolic pathways; gain a selective advantage over differentiated cells, which respond preferentially to catabolic hormones, depleting their stores for the benefit of tumor cells. The pancreatic GABA selection switch between anabolism and catabolism explains the process, a failure of GABA leads to the dual release of catabolic and anabolic hormones, and to a progressive desensitization of insulin receptors in differentiated cells. New stem cells with non-desensitized insulin receptors respond to dual anabolic and catabolic signals and rewire their metabolism in a cancer mode.
Since these mitotic cells synthesize fatty acids, there is an automatic blockade of their degradation and closure of this source of acetyl CoA, since pyruvate kinase M2 and pyruvate dehydrogenase are also inhibited, the glycolytic source acetyl CoA is also closed. Tumors will survive by converting in their mitochondria; ketone bodies into acetyl CoA, inhibiting this conversion would kill them, or force them to normalize their metabolism.
RÉFÉRENCES
- Israël M. Signaling and metabolism in cancer: endocrine pancreas deficiency and hybrid anabolism-catabolism, drugs that undo the process. Cancer Therapy. 2014;10:1-12.
- Israël M. A possible primary cause of cancer: deficient cellular interactions in endocrine pancreas. Molecular Cancer. 2012a;11: 63-68.
- Israël M. A primary cause of cancer: GABA deficiency in endocrine pancreas. Cancer Therapy. 2012b;8: 171-183.
- Israël M, Schwartz L. The metabolic advantage of tumor cells. Molecular Cancer. 2011; 10(70): 1-12.
- Braun M, Wendt A, Buchard K, Salehi A, Sewing S, Gromada J, Rorsman P. GABAB receptor activation inhibits exocytosis in rat pancreatic-β-cells by G-protein-dependent activation of calcineurin. J. Pysiol. 2004; 559(Pt2):397-409.
- Taneera J, Jin Z, Jin Y, Muhammed S.J, Zhang E, Lang S, Korsgren O, Renström E, Groop L, Birnir B. γ-Aminobutyric acid (GABA) signaling in human pancreatic islets is altered in type 2 diabetes. Diabetologia. 2012; 55(7): 1985-1994.
- Fenselau A, Wallis K. Substrate specificityand mechanism of action of acetoacetate Coenzyme A transferase from rat heart. Biochemistry. 1974; 13(19): 3884-3888.
- Pickart CM, Jencks WP. Formation of stable anhydrides from CoA transferase ans hydroxamic acids. The Journal of Biological Chemistry. 1979 ; 254(18): 9120-9129.
- Pal D, Saha S. Hydroxamic acid –Anovel molecule for anticancer therapy. J Adv Pharm Technol Res. 2012; 3(2) : 92-98.
- Madhok BM, Yeluri S, Perry SL, Huges TA, Jayne DG. Dichloroacetate induces apoptosis and cell-cycle arrest in colorectal cancer cells. Br J Cancer. 2010; 102 :1746-1752.
- Ayat M. Dichloroacetate is promising for treating hematological malignancy through inhibiting ketone bodies oxidation : towards better understanding of its cancer mechanisms. American journal of cancer prevention. 2018; 6(1):5-8.
- Israël M, Tucek S. Utilisation of acetate and pyruvate for the synthesis of ‘total’, ‘bound’ and ‘free’ acetylcholine in the electric organ of Torpedo. J Neurochem. 1974; 22(4): 487-91.
- Focke M, Feld A, Lichtenthaler HK. Allicin, a naturally occuring antibiotic from garlic, specifically inhibits acetyl-CoA Synthetase. FEBS. 1990; 261(1): 10610
13bis Bernstein BA, Richardson T, Amundsen CH. Inhibition of Cholesterol biosynthesis and Acetyl-CoA synthetase by bovine Milk and Orotic acid. Journal of Dairy Science. 1977 60(12)1846-1853.
- Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz A, Walters H, Tantawy MN, Fu A, Manning HC, Horton JD, Hammer RE, McKnight SL, Tu BP. Acetate dependence of tumors. Cell. 2014; 159(7):1591-602.
- Lux MC, Standke LC, Tan DS.Targeting adenylate-forming enzymes with designed sulfonyladenosine inhibitors. J antibiot ( Tokyo). 2019; 72 (6): 325-349.
- Koselny K, Green J, Favazzo L, Glazier VE, DiDone L, Ransford S, Krysan DJ. Antitumor/Antifungal Celecoxib derivative AR-12 is a non-nucleoside inhibitor of ANL-family adenylating enzyme acetyl CoA synthetase. ACS Infectious Diseases. 2016; 2: 268-280.
- Flavin R, Peluso S, Nguyen PL, Loda M. Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol. 2010; 6(4) : 551562.
- Israël M. Metabolic rewiring of stem cells and differentiated cells in cancer : the hypothetical consequences of a GABA Deficiency in endocrine pancreas. J Cancer Metastasis Treat. 2019; 5(12) http://dx.doi.org/10.20517/2394-4722.2018.78
- Mazurek S1, Eigenbrodt E. The tumor metabolome. Anticancer Res. (2003) ; 23: 1149-1154
Légendes
Figure 1. Conséquences métaboliques d’un déficit de la libération de GABA par les cellules bêta du pancréas
Si les cellules bêta de Langerhans, libèrent l’insuline sans parvenir à libérer suffisamment de GABA, elles ne pourront plus bloquer les libérations de glucagon ou de somatostatine, à partir des cellules alpha et delta du pancréas (seules les cellules alpha sont représentées). Cette action inhibitrice du GABA est assurée par des récepteurs ionotorpiques de type GABA A. Les cellules alpha, qui ne sont plus inhibées, vont alors libérer du glucagon, simultanément à la libération de l’insuline par les cellules Bêta. En outre, le déficit en GABA n’activera plus des autorécepteurs métabotropiques de type GABA B sur la cellule bêta, ces récepteurs permettent d’arrêter la libération d’insuline pour terminer son action. La fuite, chronique d’insuline entrainera avec le temps, une désensibilisation des récepteurs de l’insuline des cellules différenciées. Ces cellules auront une réponse essentiellement catabolique mobilisant leurs réserves. Par contre de nouvelles cellules souches dont les récepteurs à l’insuline n’ont pas été soumis au processus de désensibilisation, auront une réponse double anabolique/catabolique induite par l’insuline et le glucagon libérés simultanément en raison du déficit en GABA ; ces cellules rebrancheront autrement leur métabolisme, tel qu’il est observé dans le cancer.
Figure 2. Cétogenèse hépatique des corps cétoniques et cétolyse dans les cellules tumorales.
Nous avons représenté la bêta oxydation, dans un hépatocyte (à gauche), les acides gras sont transportés dans la mitochondrie via le transporteur à carnitine, et produiront de l’acétyl CoA intra mitochondrial. La cétogenèse est ensuite assurée par quatre enzymes indiqués sur la figure, ils formeront le Bêta hydroxy butyrate, la forme transport de l’acétoacetate, celui-ci est aussi dégradé en acétone et CO2 (non représenté). Le Béta hydroxy butyrate libéré, va être transporté dans la cellule tumorale, qui est représentée à droite. Il pénètre dans sa mitochondrie où trois enzymes vont assurer la cétolyse, aboutissant à de l’acetyl CoA, seule la succinyl CoA acetoacetate- CoA transferase (SCOT) est spécifique de la cétolyse. On voit que les autres sources d’acétyl CoA: glycolytique (via Pyruvate kinase et Pyruvate deshydrogénase) ou à partir des acides gras par bêta oxydation sont fermées, dans les cellules tumorales. Ainsi seuls les corps cétoniques via SCOT formeront de l’acetyl CoA pour alimenter le cycle de Krebs de la cellule tumorale. La réaction de condensation en citrate y est particulièrement active, le citrate va ensuite quitter la mitochondrie et alimenter la lipogenèse, indispensable pour faire les membranes des cellules tumorales mitotiques. Le blocage de SCOT de l’efflux de citrate vers le cytosol et de L’ATP citrate lyase, va freiner le développement tumoral. Dans Le cytosol nous indiquons que la cellule tumorale peut utiliser l’acétate et l’acétoacetate pour alimenter les voies de synthèse lipidiques, ces synthétases pourront aussi être inhibées. La dépendance de a cellule tumorale pour les corps cétonique, la rend vulnérable à l’inhibition de SCOT, ou elle pourrait être contrainte de normaliser son métabolisme.
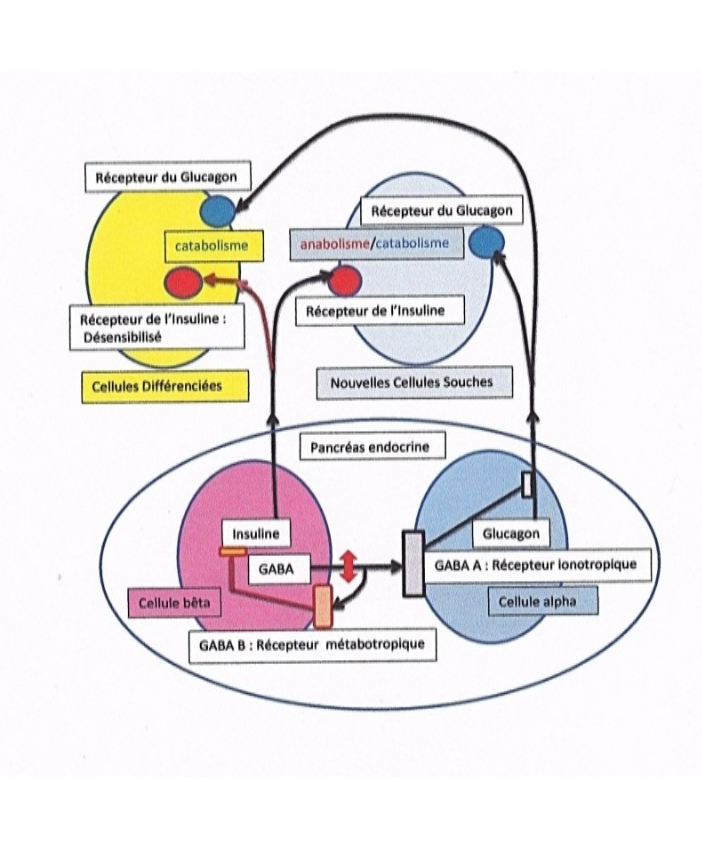
Figure 1
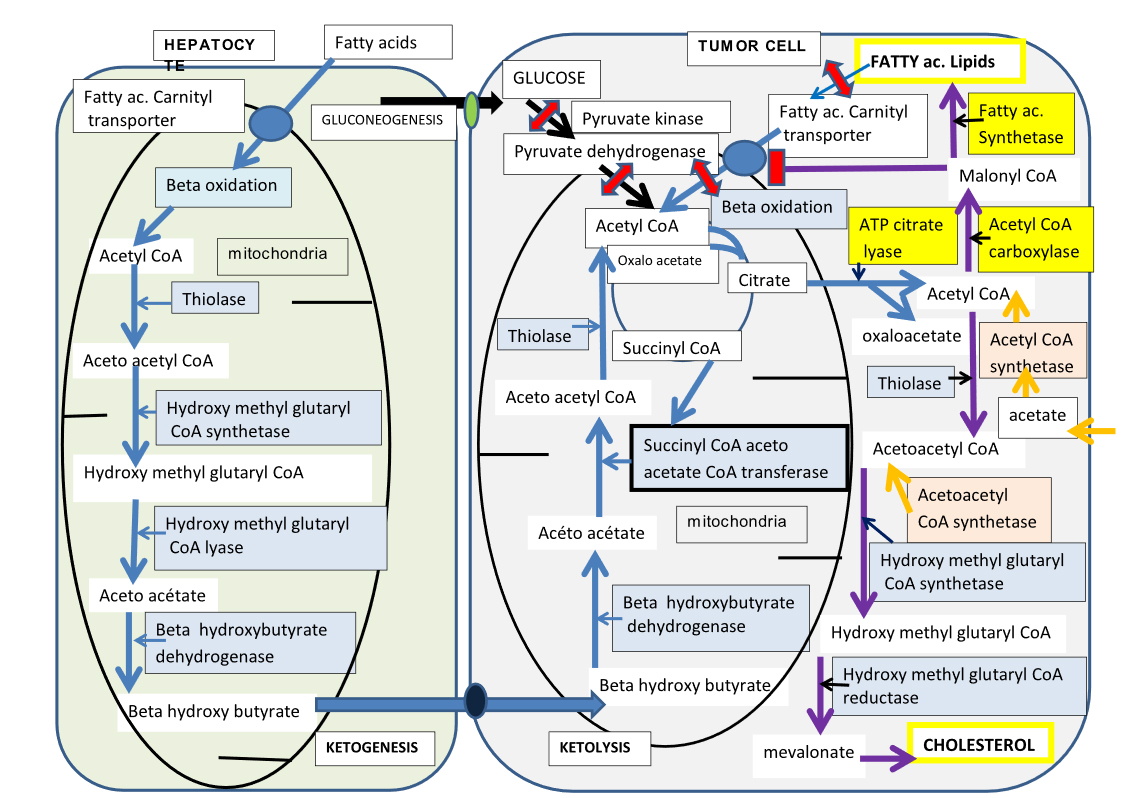
Figure 2
Addendum et incertitudes.
À propos du régime cétogène et cancer.
Dans une publication récente, RG. Klement (Journal of traditional and Complementary Medicine
9 (2019) : 182-200) retrace l’historique du régime cétogène dans le Cancer. On y trouve quelques observations indiquant des effets positifs sur certains cancers ; mais de nombreuse publications montrent une aggravation due aux corps cétoniques (Martinez –Outschoorn et.al Cell cycle 2012 11 :21,3964-3971). Ce que fait remarquer Klement dans son article c’est que la première utilisation de ce régime dans le cancer est attribuable au Dr Wilhelm Brünings 1941. Ce médecin connaissait les travaux de Warburg sur l’utilisation du glucose par la tumeur avec production de lactate même sous oxygène. Brüning a alors pensé qu’il fallait priver la tumeur de glucose en provoquant une hypoglycémie, pour cela il a utilisé l’insuline, associée à régime pauvre en sucre et riche en corps gras, le régime cétogène. Une observation initiale sur un patient diabétique portant une tumeur maxillaire fut positive, la tumeur avait régresse sous forme d’une masse gélatineuse comme il le constate lors de son ablation chirurgicale. Mais les études qui suivirent ont donné des résultats décevants après un début encourageant avec une stabilisation ou régression partielles des tumeurs durant les 2 premières semaines, un rebond dans les 2 ou 3 mois suivants fut décevant De nombreuses publications ont depuis montré une aggravation tumorale attribuée aux corps cétoniques (Martinez –Outschoorn et.al Cell cycle 2012 11 :21,3964-3971) .
A notre avis, le régime cétogène n’est pas une bonne idée, si l’on considère que les corps cétoniques sont l’unique source d’Acétyl CoA mitochondrial pour les cellules tumorales, et qu’ils assureront alors leur développement. Il faut aussi remarquer que le régime cétogène n’est donné que pour remplacer, d’un point de vue alimentaire, le manque de sucre lié au traitement hypoglycémie par de l’insuline, dont l’objectif était de réduire l’apport de glucose à la tumeur.
Chez un Diabétique de type I le manque d’insuline, élève le glucose sanguin et provoque une élévation des corps cétoniques. Or la cétogenèse est en général provoquée par l’action du glucagon, la baisse d’insuline chez le diabétique pourrait lever l’effet inhibiteur de l’insuline sur la sécrétion du glucagon, qui va alors être libéré et déclencher la synthèse des corps cétoniques et une production de glucose aggravant l’hyperglycémie due au déficit d’insuline.
A l’inverse, si l’on injecte de l’insuline à un sujet sain, on provoquera une hypoglycémie, mais aussi une inhibition par l’insuline, de la libération de glucagon, qui ne formera ni glucose pour compenser l’hypoglycémie insulinique, ni corps cétoniques. Au contraire, l’hypoglycémie du jeûne, non insulinique, agira directement ou indirectement sur la cellule alpha, pour déclencher une libération de glucagon induisant la production de glucose et la cétogenèse.
Revenons au Cancer et à l’injection d’insuline, elle provoquera une hypoglycémie avec inhibition de la libération de glucagon, ce blocage du glucagon ne corrigera pas l’hypoglycémie provoquée par l’insuline, et ne produira pas des corps cétoniques, les injections d’insuline seule, devraient alors bloquer le développement de la tumeur ; privée de corps cétoniques et de glucose. C’est bien ce qui a été observé par Koroljow1962 (Cité dans l’article de Klement 2019). Mais si l’on rajoute à l’hypoglycémie insulinique le régime cétogène, il y aura ambiguïté des résultats, et reprise du développement tumoral, puisque la tumeur est dépendante, comme nous l’avons vu, de cette seule source d’Acétyl CoA mitochondrial. C’est probablement l’hypoglycémie qui améliore l’évolution de certains cancers, mais lorsque l’on utilise l’insuline pour la provoquer, on court aussi le risque d’induire sur sa voie de signalisation une élévation de protéines qui à chacune des étapes sont les principaux oncogènes cellulaires. Baisser la glycémie autrement que par l’insuline (elle active les mitoses et les oncogènes cellulaires), mais en en inhibant la libération de glucagon, pour ne pas produire les corps cétoniques indispensables pour la tumeur, pourrait avoir un effet anti-tumoral, mais comment faire ?


Article très intéressant, merci